
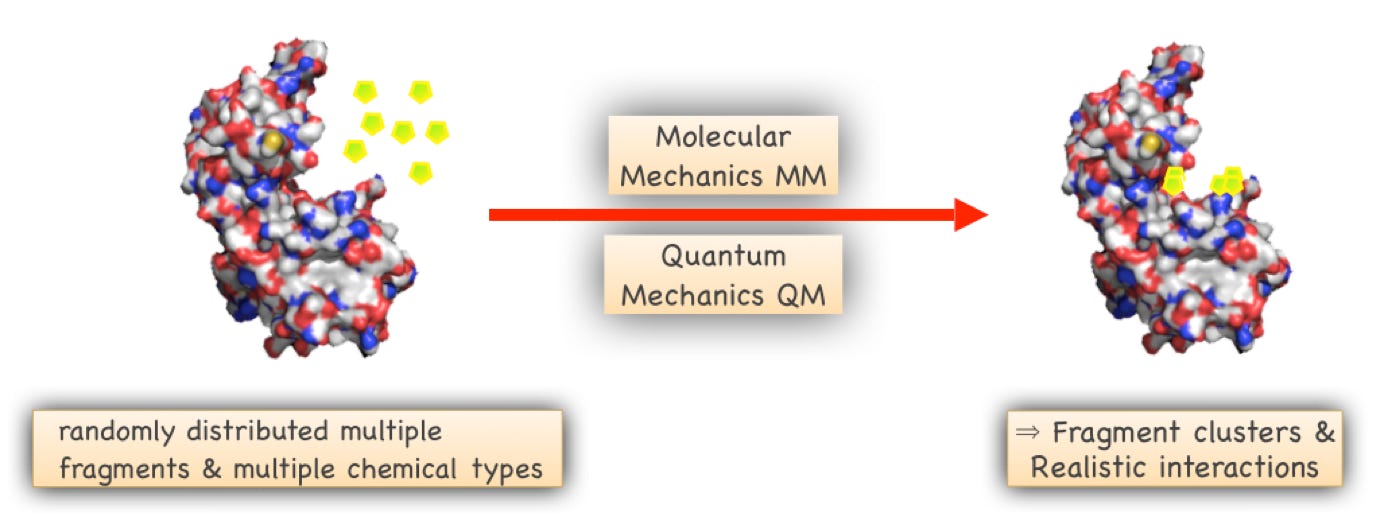
MFMD is based on the “MCSS” method we developed for computational design of protein-protein inhibitors and conventional target binding sites. The origin of the method is published in: Zeng J. and Treutlein HR, A method for computational combinatorial peptide design of inhibitors of Ras. Protein Engineering 12, 457-468, 1999.
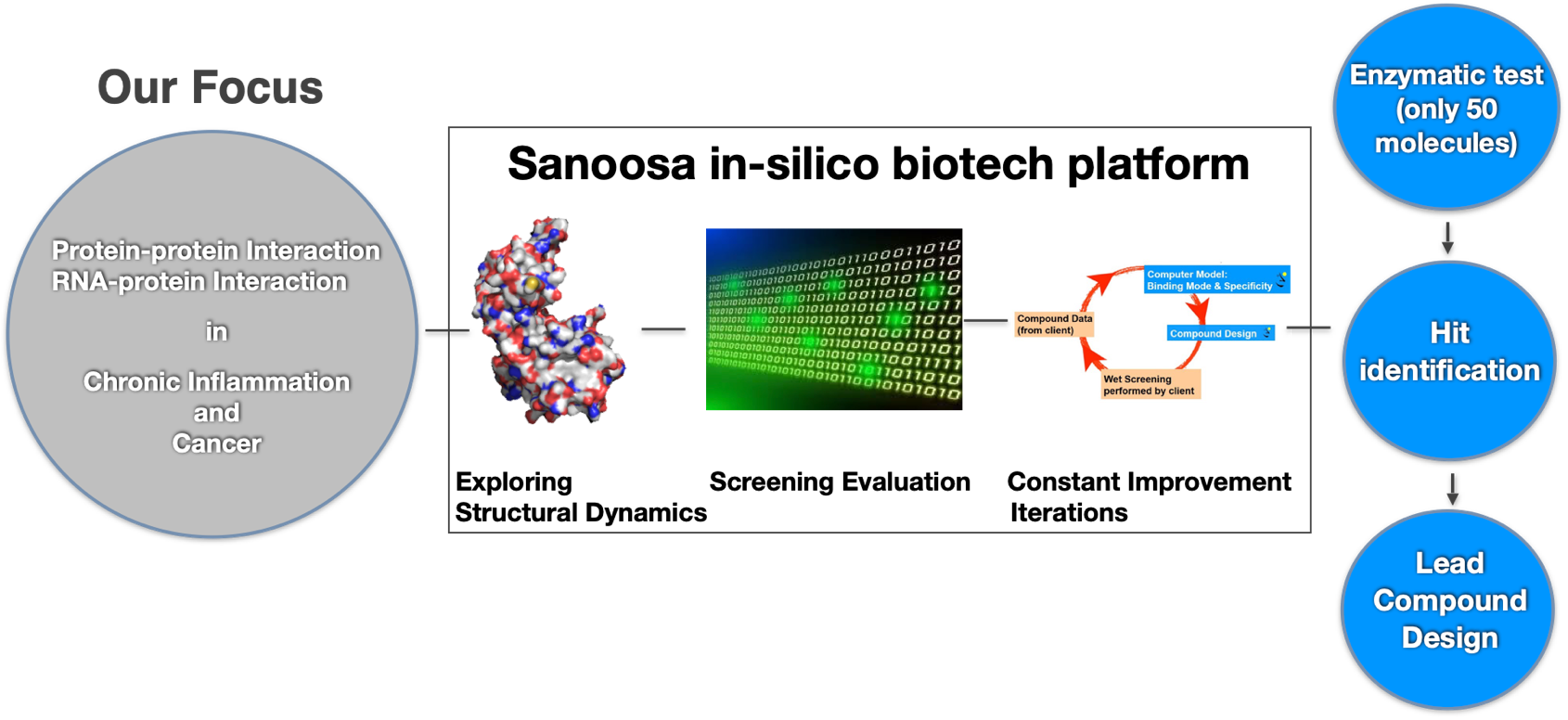
Our “Multiple Fragment Molecular Dynamics” (MFMD) scans of a target binding pocket not only tell us whether compounds are well suited to a target, but also indicate ways on how to improve the compounds.
Our outcomes speak for themselves. Using MFMD we have successfully predicted ligand binding modes and optimised inhibitors for numerous targets in collaborations with both members of academia and partners in the pharmaceutical industry. Click here for references to our published projects.
This website makes use of cookies. Please see our privacy policy for details.
This website makes use of cookies. Please see our privacy policy for details.